Specification list
On this page, are described BioFlow-Insight’s Functionalities and Guidelines.
Table of Contents
BioFlow-Insight’s Functionalities
Workflow structure reconstruction
Description
Nextflow workflows are based on the dataflow programming model, wherein processes, encapsulating specific bioinformatics tasks using scripts or tools, communicate through channels—either non-blocking unidirectional FIFO queues or single values. Nextflow operators are methods that allow users to manipulate channels, such as filtering, forking, or maths operators. For more information on Nextflow workflows, please refer to Nextflow’s official documentation. Additionally, multiple operators can be associated; we define this association as forming a single operation.
Multiple representations of the workflow are generated by BioFlow-Insight representing it at different levels of granularity.
-
Specification graph: BioFlow-Insight reconstructs the workflow’s specification graph from its source code without having to execute it. The specification graph is defined as a directed graph where nodes are processes and operations, and edges are channels that are directed from one vertex to another (steps of the workflow are ordered). This graph represents all the possible interactions between processes and operations through channels that are defined in the workflow code. Within the specification graph, we define two types of operations: operations are categorised in two groups: the following operations defined as operations that have at least one input, and the starting operations defined as operations without any inputs.
-
Dependency graph: From the specification graph, BioFlow-Insight also generates the dependency graph which represents starting operations, along with processes (as nodes) and their dependencies (edges). This graph is obtained by removing the following operations and linking the remaining elements if a path exists between them in the original specification graph. In this representation, the edges no longer represent interaction between its elements, but their dependencies.
-
Process dependency graph: Finally BioFlow-Insight also generates the process dependency graph which represents only processes (nodes) and their dependencies (edges). Similar to the dependency graph, this graph is constructed by removing all operations, leaving only processes, and linking them based on their dependencies in the original specification graph. Again in this representation, the edges no longer represent interaction between its elements, but their dependencies.
Here are the 3 structures generated by BioFlow-Insight from the https://github.com/George-Marchment/hackathon workflow.
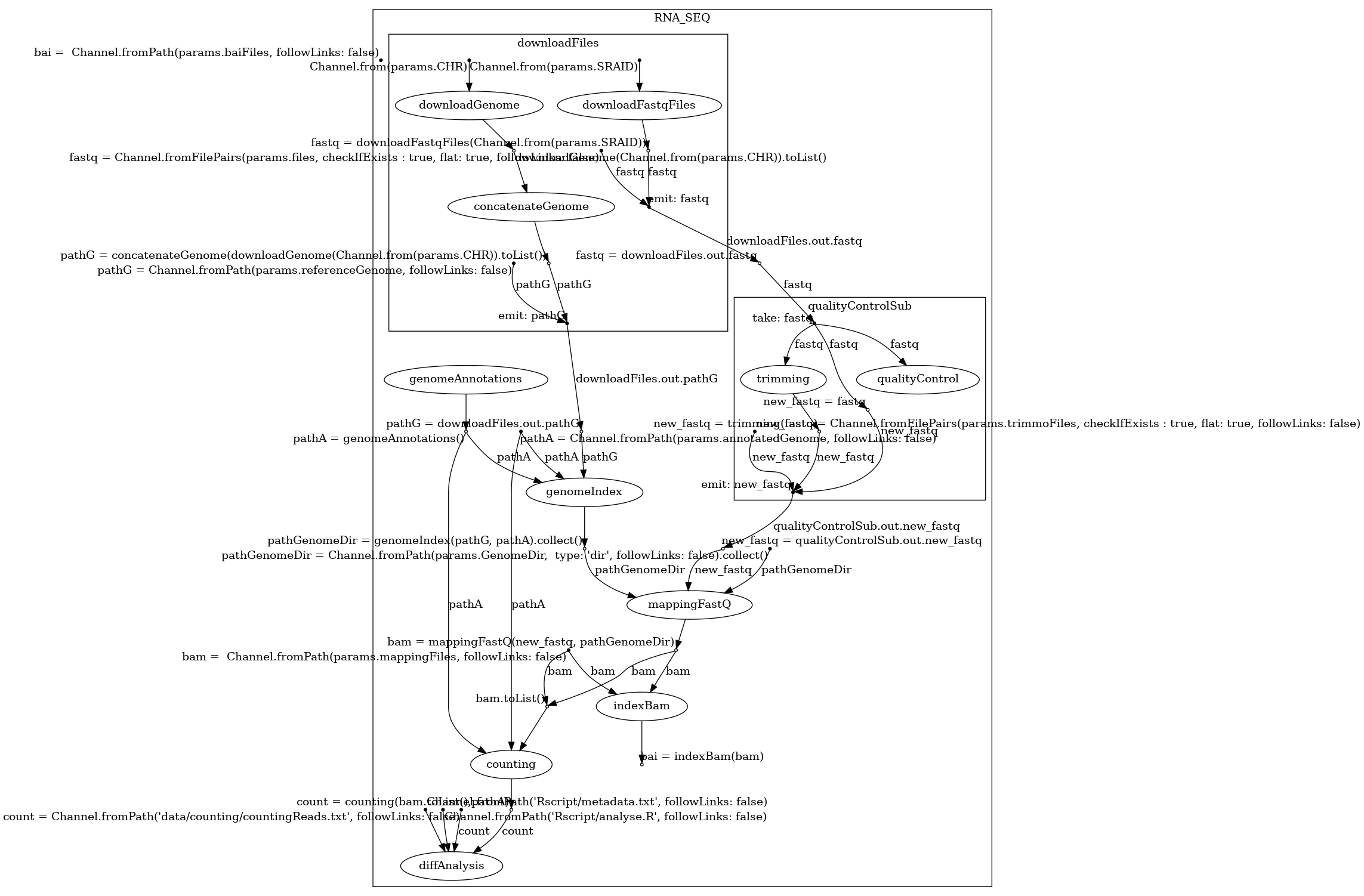
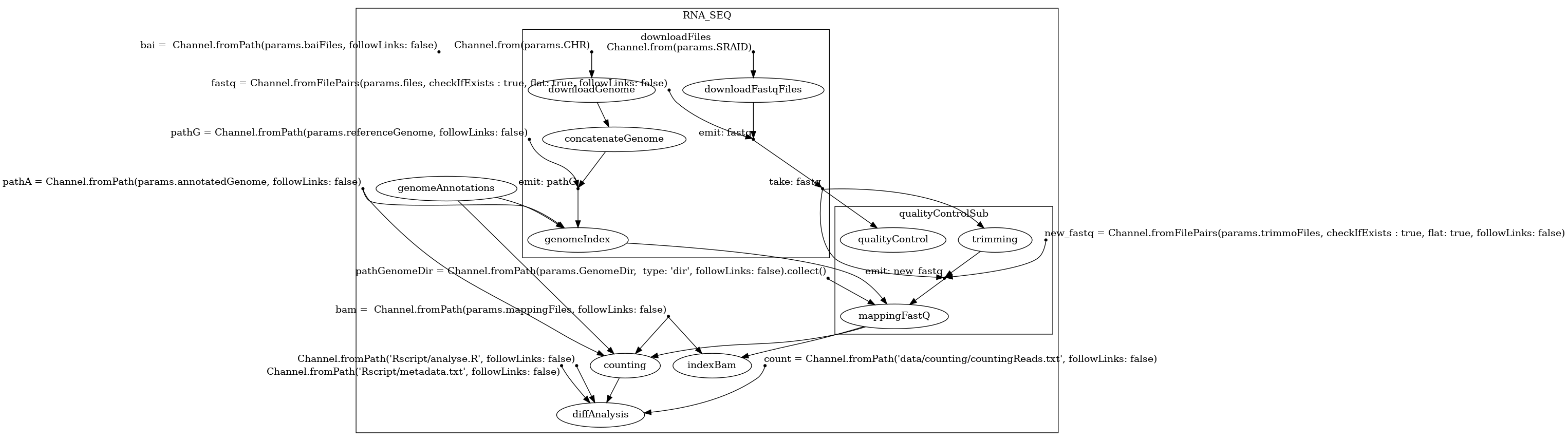
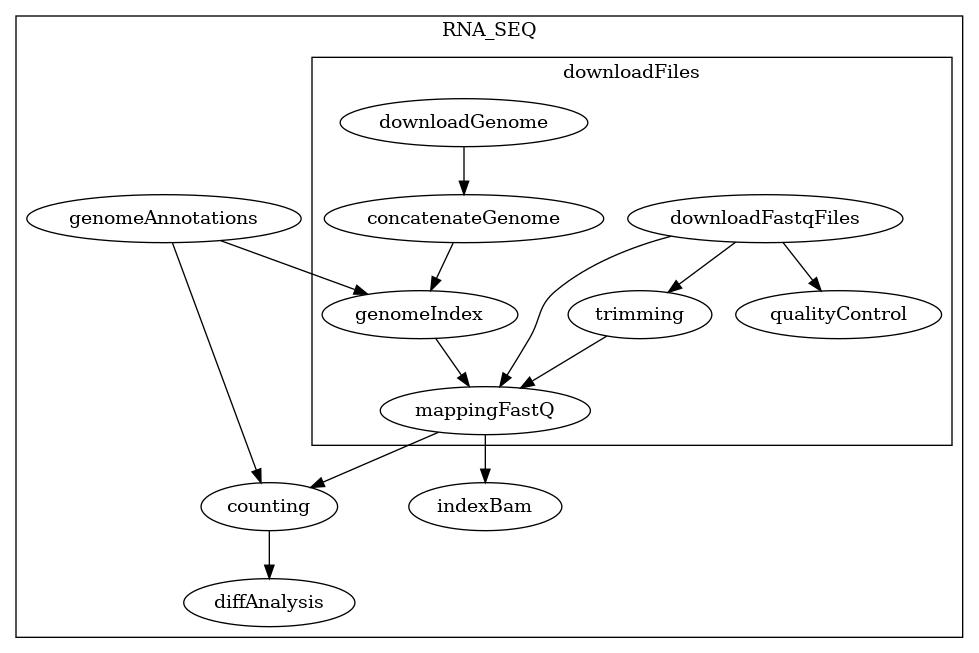
The specification graph on the left, the dependency graph in the middle and the process dependency graph on the right.
Labelled and unlabeled versions of the graphs are also available. With variants where orphan operations (operations without any inputs or outputs) are represented or removed.
For large workflows, it can be useful to represent only a subset of its processes, by excluding certain ones. This is why BioFlow-Insight provides the option to remove a list of specified processes from the representations. By default, this list is empty, meaning that all processes are represented.
BioFlow-Insight can analyse both DSL1 and DSL2 Nextflow workflows.
Examples
To test BioFlow-Insight’s structure reconstruction, try these example workflows with the 'Submit from a git repository' functionality (add link):
- https://github.com/nf-core/phyloplace
- https://github.com/George-Marchment/hackathon
- https://github.com/maxemil/ALE-pipeline
- https://github.com/x-kiana/nextflow_pipeline
Workflow error detection and handling
BioFlow-Insight analyses Nextflow workflows and code. It is by no means a Nextflow "checker" or "validator". Therefore, while BioFlow-Insight can analyse a workflow, it does not guarantee its functionality during execution. However, it serves as a helpful tool for Nextflow workflow developers to identify some errors in their workflow’s code.
When BioFlow-Insight fails to analyse and generate structures, this can occur for two reasons.
- The first reason is that there is an error or ambiguity in the workflow's source code.
- The second reason is that the workflow may be written in a manner that BioFlow-Insight, due to its limited scope, does not know yet how to handle—because certain code instances are not yet considered.
Both of these cases are elaborated upon below.
Errors or ambiguities detected in the workflow’s code
Description
Here is an extensive list of errors and ambiguities in a Nextflow workflow detected by BioFlow-Insight:
- Not the right number of parameters given for a process or a subworkflow when calling them.
- A channel is trying to be created with a name already given to something else in the workflow (e.g., a process or a subworkflow).
- Multiple channels were given by an emitted value even though only one was expected (e.g., in an operation).
- Tried to access an emitted value even though the element from which it's being emitted has not been called (e.g., a process or a subworkflow).
- Tried to include a code file which doesn't exist.
- An include is present in a main or subworkflow section.
- In a pipe operator, the first element being called is unknown (e.g. it has not been included or it is not defined).
- General syntax errors in the code (detecting unclosed parentheses or unclosed quotes).
- An element is expected to be defined (e.g., a process or subworkflow) in an included file but it is not.
- When using an emitted value from a subworkflow in an operation, if the subworkflow emits nothing or too many values.
- A subworkflow or process was poorly defined (e.g., multiple identical tags in a process such as "input" or "output").
Examples
To test BioFlow-Insight’s error detection, try these example workflows with the 'Submit from a git repository' functionality:
- https://github.com/George-Marchment/NF-IAP (missing file in the workflow)
- https://github.com/George-Marchment/jasen (channel is trying to be created with a name already given to a process)
- https://github.com/George-Marchment/chipseq (Tried to access an emitted value even though the element from which it's being emitted has not been called)
BioFlow-Insight’s limited scope
Due to Nextflow's highly flexible workflow definition, there are some limited cases where BioFlow-Insight cannot analyse the workflow’s code. Below is a list of these cases:
- In a pipe operator when wanting to associate multiple processes and subworkflows, BioFlow-Insight cannot interpret a call of type
ch | (process1 & process2 & process3). The call would have to be rewritten asch | process1 | process2 | process3orch | process3 | process2 | process1depending on the ordering wanted. - The use of a tuple in association with a ternary conditional operator, for example the syntax
(ch1, ch2) = (condition ? [val1_1, val2_1] : [val1_2, val2_2])is not supported by BioFlow-Insight. The operation would need to be decomposed into 2 operationsch1 = condition ? val1_1 : val1_2andch2 = condition ? val2_1 : val2_2. - The association of a tuple with an emit, for example the syntax
(ch1, ch2) = emit.outis not supported by BioFlow-Insight. The operation would need to be decomposed into 2 operationsch1 = emit.out[0]andch2 = emit.out[1].
Important: If BioFlow-Insight fails to analyse a workflow due to its limited scope, it is easy to rewrite the workflow in a different way to enable successful analysis. When possible BioFlow-Insight also specifies the reason it failed.
Metadata extraction
After the extraction of the graphs, BioFlow-Insight analyses each graph and extracts a certain amount of metadata. Below are the attributes that are calculated:
- Number of processes
- Number of operations
- Number of nodes
- Number of edges from process to process
- Number of edges from process to operation
- Number of edges from operation to process
- Number of edges from operation to operation
- Number of edges from source process
- Number of edges from source operation
- Number of edges to sink process
- Number of edges to sink operation
- Total number of edges
- Number of simple loops (nodes connected to themselves)
- Distribution, average, median for in-degree and out-degree for processes and operations
- Density
- Number of weakly connected components
- Number of weakly connected components with two or more nodes
- Number of cycles
- Structure type (either a directed acyclic graph or a cyclic directed graph)
- Number of paths from source to sink (by rooting the graph and ignoring cycles)
- Shortest path (by rooting the graph)
- Longest path (by rooting the graph and ignoring cycles)
These metadata are saved in dedicated JSON files.
To obtain the rooted graph, we add 2 nodes: the source and the sink. The source is connected to all nodes which do not have any incoming edges (\(indegree=0\)). The sink is linked to all nodes that do not have any outgoing edges (\(outdegree=0\)).
RO-Crate generation
BioFlow-Insight generates a description of the workflow in the RO-Crate format. RO-Crate serves as a standard for aggregating and describing research data, including associated metadata for workflows and scripts. However, the current framework of RO-Crate does not yet fully accommodate Nextflow workflows. For instance, in the current RO-Crate format one subworkflow is equal to one file. To address this limitation, BioFlow-Insight extends the RO-Crate framework. This extension also enables a comprehensive description of Snakemake workflows. For a description of this extended profile, check-out its description which can be found here: https://gitlab.liris.cnrs.fr/sharefair/posters/swat4hcls-2024.
When analysing from a workflow from GitHub repository, relevant metadata such as authors, keywords and the last update are automatically extracted.
BioFlow-Insight’s Guidelines
Due to the highly flexible nature of Nextflow's workflow definition, BioFlow-Insight may not handle certain code syntaxes. This section provides guidelines for defining your workflow to ensure it can be effectively analysed by BioFlow-Insight. Additionally, for cases not handled by BioFlow-Insight, easy alternatives are provided.
For full information on how to define a Nextflow workflow, please refer to the Nextflow documentation. It is recommended to follow the recommended syntaxe given by Nextflow.
General guidelines:
- The Nextflow files which are being analysed need to be a hard copy of the file and not a shortcut redirecting to a different address.
- For a DSL2 workflow, there needs to be a main i.e. the entry point of the workflow.
Below Syntaxe and Functional guidelines are provided.
Syntaxe Guidelines
- Same number of opening and closing brackets in all the Nextflow files composing the workflow.
- Same number of opening and closing parentheses in all the Nextflow files composing the workflow.
- When a quote
‘or“is opened, it needs to be closed. - When using quotes to define a string please use the standard quote symbols which are
“...”and‘...’ - In DSL2 a process, subworkflow or function cannot be called using a bracket (“{}”). For example,
element{ch1, ch2}becomeselement(ch1, ch2) - When defining a variable or a channel, use standard attribution syntax i.e.
ch = valueand notch << valueorch <- value - Channels, processes or subworkflows should not be given a name from this list:
["null", "params", "log", "workflow", "it", "config", "channel", "Channel", "null", "params", "logs", "workflow", "log", "false", "true", "False", "True", "it", "config"]. - Channels cannot be defined with the name of existing processes or subworkflows.
- Channels cannot be defined with the name of existing operators:
["distinct", "filter", "first", "last", "randomSample", "take", "unique", "until", "buffer", "collate", "collect", "flatten", "flatMap", "groupBy", "groupTuple", "map", "reduce", "toList", "toSortedList", "transpose", "splitCsv", "splitFasta", "splitFastq", "splitText", "cross", "collectFile", "combine", "concat", "join", "merge", "mix", "phase", "spread", "tap", "branch", "choice", "multiMap", "into", "separate", "tap", "count", "countBy", "min", "max", "sum", "toInteger", "close", "dump", "ifEmpty", "print", "println", "set", "view", "empty", "of", "fromPath", "fromList", "subscribe", "value", "from"]. See the list of operators here. - Channels should not be defined with the same name as the file in which it is defined.
- Avoid associating the ternary conditional operator in a ternary conditional operator, for example:
ch1 = condition1 ? condition2 ? val2_1 : val2_2 : val1, needs to be rewritten asif(condition1){if(condition2){ch1 = val2_1} else {ch1 = val2_2}} else {ch1 = val1}. - Avoid associating the ternary conditional with new lines (
\n). - When using the ternary conditional operator do not use a tuple for example:
(ch1, ch2) = (condition ? [val1_1, val2_1]: [val1_2, val2_2])needs to be rewritten asch1 = condition ? val1_1: val1_2andch2 = condition ? val2_1: val2_2`` Cannot call emitted values directly from a call, for example:process().outneeds to be rewritten as firstprocess()and then useprocess.out`. - Includes need to be defined outside of main and subworkflows.
- A tuple cannot take the value of an emit, e.g.
(ch1, ch2) = subworkflow.out
Functional Guidelines
- When defining a call to a process, subworkflow or function; the element which is being called needs to be defined prior to its call. For example, for the call
element(ch1)thenelementneeds to be defined in the file prior to the call. - In a pipe operator when wanting to associate multiple processes and subworkflows, do not write
ch | (process1 & process2 & process3)but writech | process1 | process2 | process3orch | process3 | process2 | process1depending on the ordering wanted - When defining a call it needs to be inside either the definition of the main or a subworkflow.
- In subworkflows, at most one section for take, emit and main can be defined.
- In processes, at most one section for inputs and outputs can be defined.
- When using the emitted channel of a subworkflow as such
subworkflow.outthe subworkflow should only emit one single channel. If multiple channels are emitted, then the channel needs to be selected either bysubworkflow.out.chorsubworkflow.out[0] - When defining a call, the correct number of inputs needs to be given.